I think the above objection is an excellent one. In it’s favour, we observe whole phenotypes that converge to a high degree of uniformity (i.e., wild-type dog) when the full range of functional whole phenotypes might be vastly broader (i.e., the enormous range of domestic dogs). As for protein families, if we assume their fitness gradients resemble a hill with a global maximum, it is easy to write a genetic algorithm with an arbitrary functional cut-off “E(x)” and demonstrate that it will converge on a small subset of sequences near the top of the global maximum much smaller in sequence space than the larger functional set.
Response: The data does not support this hill-climbing view for the fitness gradients of protein families.
a) There is an increasing recognition in biology that the bulk of evolution is not explained by natural selection but by genetic drift and neutral mutations and epistasis. More specifically, the fitness gradients are often insufficient to make any selectable difference.
b) We observe genes/proteins that teeter on the edge of functionality but are still retained in the human genome and show no signs of disappearing due to purifying selection. Two examples are haemoglobin S and transthyretin (which is the reason I included it in a subsequent paper). Furthermore, with epistasis (‘Epistasis as the primary factor in molecular evolution’, Nature, Vol 490, October 2012) we observe that some alleles are non-functional in some genetic backgrounds and perfectly functional in others. Thus, sampling across large numbers of taxa, we can find a lot of alleles at the edge of functionality.
c) Most importantly , the data does not support the hill-climbing fitness gradient theory. A “mesa” would be a more apt description, with relatively steep fitness gradients at the boundary of functional sequence space, and a relatively low-relief top representing functional sequence space. I’ve written a program to analyze in more detail the multiple sequence alignments in Pfam. What I find from the actual data is that protein families show an average of 15 to 18 different amino acids per site. Ubiquitin, for example, shows an average of 18 different amino acids per site (11,656 unique sequences analyzed). SecY shows an average of 16 aa’s/site (5714 unique sequences analyzed). I’m not cherry picking here; of the 31 universal proteins I’ve looked at in the last few months, the most highly conserved one was chorismate synthase, showing an average of 14 different aa’s/site for 4,457 unique sequences. I realize that pairwise dependencies and epistasis make it such that there cannot be a uniform probability distribution for the different aa’s/site, but from these data, there is a way to estimate an extreme lower limit for functional information required to code for a protein family.
Great. It seems we have identified a process that, if real, you agree would make FSC (your measure) an inaccurately high measure of FI. Great.
You have also, correctly, identified FSC as a special case of Algorithmic Specified Complexity (ASC). Marks argues that ASC is guaranteed to never overestimate FI. You are now in disagreement with him. You are joining @EricMH in this disagreement with Marks. I’m glad to hear this.
This is a very surprising claim. Do you really believe this?
It turns out that even Behe, Axe, Gauger, and most ID advocates all agree that once a function appears, it can be optimized. Axe, for example, argues that the issue is just the finding the function to optimize is the problem. Once it appears, we have ample evidence that evolution can optimize it. Is it really your intention to disagree with them on this?
This just shows that we do not need a direct path to optimize function. This undermines your claim.
This is essentially a bad design argument. Is it seriously your argument that the human body is not well optimized? Often proteins in humans are optimized to be operating at a balance of different factors. Too much activity and they create problems, to little and the do not create problems. Also many of them are unnecessary any ways, we know of about 1300 natural knock outs with no apparent phenotypes. They are all likely neutral changes.
It also does not make your case. The proteins you’ve focused on are ancient, and ubiquitous. Those proteins surely are optimized. You’d have to demonstrate for each case that evolution had no ability to optimize that family before we could trust an FSC measurement on that family. Good luck.
Once again, you are in disagreement with most ID leaders on this, who acknowledge the immense amount of data that evolution can optimize protein function. They way they have argued against this is to insist that getting minimum function is vanishingly improbable. You are taking a different approach here, arguing against them that evolution cannot even optimize protein function. Why do you disagree with them?
I am cautious about saying I disagree or agree with anyone if I have not carefully read their work. Consequently, I cannot comment on Marks’ approach. In general, I have no concern for who I agree or disagree with, ID or no ID. I am much more interested in what the data actually shows and with the proper way to analyze the data. For clarity, here is a summary of what I have laid out thus far:
The definition of information gain since Brillouin, and subsequently known as the Kullback-Leibler information gain, is simply the difference between the Shannon information of two states. This is an established definition that has been around for over 60 years.
The K-L information gain should not be used as a distance metric, since it is not symmetric (as you correctly mentioned in one of your comments). It is simply a measure of information gain (or loss). It can be positive (information gain) or negative (information loss).
The definition of functional information that I use, as well as Hazen, Szostak, et al. is the K-L information gain between two states where one state is non-functional and the other is functional. I don’t see anything controversial here; I’m merely defining the properties of the two states I’m interested in.
Most, if not all, the concerns you have with the K-L approach to functional information has to do with a) defining states A and B and b) the problem of sampling the two states.
In theory, Algorithmic Complexity (AC) should never overestimate information (functional or otherwise) but in reality, the problem is knowing whether the information string is the shortest possible for the given function. I don’t know how Marks determines the shortest string for protein families, so I can’t comment on this, but even if he has a good method, he must ultimately (as does AC in general) use K-L information to quantify that shortest string in terms of bits. I do significantly pair down the sequences in the Pfam MSA’s to what I observe (or my program “observes”) to be the shortest sequences the cell will permit, acknowledging that epistasis (closely coupled with designed optimization) may introduce some variation there. In doing this, therefore, there is a methodological relationship between AC and K-L information gain in practical application, but AC in general has some other issues.
When I say that the data does not support a hill-climbing view for the fitness gradient within functional sequence space, this is not a claim but an observation from the data. There is a significant difference between a claim and an observation. Conversely, if you or Behe, Axe, etc. believe that functional sequence space for protein families has been optimized to a small subset of functional sequence space, I would like to see the data that supports that. I think the data is on my side, and overwhelmingly so. If you wish, I can attach detailed data to support this, as well as the observation that the extreme minimum number of mutational events for universal proteins is, on average, at least 26 mutational events/site and almost certainly many times higher than that.
To be charitable, I think a lot of biologists tend to think the functional sequences have been optimized because they often look at a taxonomic subset (which I quite happily suspect might only represent a small subset of functional sequence space for the entire protein family due to epistatic requirements or, more often, simply the outcome of population dynamics and neutral evolution). This is why I would argue that sampling discrete taxa or related taxa may result in grossly inadequate sampling. My work at present is focusing on the sampling problem, something that is common to all scientific measurements and estimations. How do we ensure we have an adequate sampling of total space? What methods can we use? How can we test the quality of our sample?
Finally, I must correct your perception that I said that evolution cannot optimize protein function. That is not true. What I said is that the data reveals that the overall fitness gradients inside functional sequence space falsifies the idea of a global maximum that forces evolution to converge on a small, unrepresentative subset of the overall functional sequence space for that family of proteins. There are other things you assume I concede that I would like to correct, but we can deal with those as they resurface.
As I said earlier, I suspect your concerns fall into the category of defining and sampling the two states. A good test will be your cancer paper, which I have not yet read, so I am nicely setting myself up here for a test of my approach.
This is so dense with mathematical errors it is hard to know where to begin.
This is not true. The way you defined KL is delta H. This is not the definition. Period. I encourage you to go back and check your references.
This is false. KL is often used as a distance metric between probability distributions. It is not a metric distance, but in many applications we are happy to use no-metric distances. As one example of literally thousands of papers that do this, look at this: [PDF] Using Kullback-Leibler Distance for Text Categorization | Semantic Scholar
KL gives a way of computing the number of bits to move from one states to the other, so it is precisely what we should be computing. For some simplistic background states, this reduces to your forumula. For others it does not.
This is both false, and the second time I’ve corrected you on this. Let me give you some quotes:
This is the Gibbs inequality and is included in all information theory textbooks. Gibbs' inequality - Wikipedia I’m fairly stunned that you did not know this, and when I corrected the error the first time, you just ignored it. Just looking in wikipedia would have shown you who was correct.
That is not their definition. You are equivocating your formula with theirs.
Given that this is the beginning of the false equivalences, I’d say it is pretty controversial.
I could get into the rest, but I’m not sure what it matters right now. The error on KL is just such a fundamental misunderstanding of information theory that nothing else is going to make sense from here until it is corrected.
My concerns are far broader than than this.
The post is available on the forum whenever you want to engage it.
Note that it is guaranteed that D_{KL} is greater than zero because the information content of two objects is always greater than or equal to the information content of either one of them alone.
H(P,Q) \geq H(P)
I hope it is very clear now that D_{KL} is not merely \Delta H. As I’ve explained already, in some cases, for example where there is no correlation between P and Q and P is higher entropy than Q, then this might reduce down to delta H. However, it is a grand error to see this as generally true.
The only reason it works this way in @kirks specific model is because of the a poor choice of a base state (IID MaxEnt). With a better choice of base state, this equivalence collapses into error.
A technical error here due to similarity of notation between cross entropy and joint entropy. While it is true cross entropy is higher (Gibb’s inequality), it is not the same as joint entropy.
Per your more general point regarding @Kirk’s errors, you are correct he is wording things incorrectly, i.e. KL divergence cannot be negative. But you should refer to the equation in his paper:
ΔH(Xø(ti), Xf(tj)) = log(W)-H(Xf(tj))
which does turn out to be a KL divergence as I explained in the UD comment you read, since log(W) is maximum entropy for X. So, no fundamental error in the math. Best to read an interlocutor’s comments in the best possible light (which I’m guilty of not doing with your comments, again apologies).
Anyways, I am jumping in here because I want to move things forward to the validity of the approach, i.e. measuring functional information as the KL from the uniform distribution. In other words, can you explain the error in assuming maximum entropy you mention:
First of all, you are correcting me on a point that I already acknowledge.
“this way” is Kirk’s claim that delta H is KL divergence (it is not). This leads him to several errors in reasoning. These are not merely misstatements. All these claims are false:
All these claims are false. Every one of them, demonstrably so. Some of them I have already told him once before. Rather than correcting the error and calling it a misstatement (which would have been great), he doubled down and repeated some of the error. They are all traceable to the same mistake in understanding. If he merely misworded this, then he wouldn’t have disagreed with me when I said this:
This is not just nitpicking. It appears that @Kirk thinks that KL can be negative and that it represents the information gain or loss between two states. This indicates he misunderstands something foundational things in information theory. If we can’t even agree that KL distance is always positive, we are not inhabiting the same mathematical universe.
Post 7: It appears we are not in 100% agreement, so I would like to pinpoint where we depart from common ground. For precision and clarity, let’s do this in a stepwise fashion, beginning with Shannon entropy and proceeding from there.
Given:H = - K ∑ p ( i ) log p ( i ) which, according to Shannon is a measure of “information, choice and uncertainty.” I trust we are in agreement thus far.
H will always be positive. I expect we have agreement here as well.
If H (A) is the Shannon information of state A, and H (B) is the Shannon information of state B, then the difference in Shannon information between the two states is H (A) – H (B). It is at this point that I am not 100% sure we are in agreement. If not, I would like to understand why you think that the difference between H (A) and H (B) is not, in fact, the difference between the Shannon information of the two states.
If H (A) is positive and H (B) is positive, and if H (A) > H (B), then H (A) – H (B) will be positive. This shows that there is an information gain between states A and B. However, if H (A) < H (B), then H (A) – H (B) will be negative. This shows that there is an information loss between states A and B.
It is here that our common ground seems to have completely vanished, so let me clarify with a very simple example.
Given: Let us imagine that site x in P53 tolerates only one amino acid out of the 20 normal possibilities. The Shannon information of that site, for a functional P53 protein is H (func) = 0 bits (in agreement with Shannon’s point (1) on page 11 of his paper). Now let us imagine an analysis of a cancerous tumour has revealed that, for the cancerous cells, P53 permits the other 19 amino acids equally well (i.e., makes no difference at all to the cancerous cell), but not the original, highly conserved one (which would terminate the cell). The Shannon information of that site, for P53 proteins that permit the cancerous cell to multiply is H (cancerous) = 4.2 bits. The change in Shannon information between the functional P53 state and the cancerous P53 state is H (func) – H (cancerous) = 0 bits – 4.2 bits = - 4.2 bits. We can conclude, therefore, that the change in state is negative, corresponding to a loss of Shannon information between the healthy cell and the cancerous cell.
Let’s pause here for discussion, as your comments suggest that we may have departed common ground at some point between (1) and (4).
Tangential to (4) Re. the K-L information gain: I have been using the K-L information gain as defined here https://en.wikipedia.org/wiki/Information_theory You will note that it is defined as the difference in Shannon information between two probability distributions, representing two states p ( X ) and q ( X ). If the Shannon information of the aposteriori state q (X) is greater than the apriori state, we have a problem with that definition if the K-L information gain must always be positive. I have no vested interest in using that definition; it seems to be more of a hindrance than an asset in this discussion, especially if we want to estimate/measure information loss. We can quite nicely define the gain (or loss) of functional information solely from the change in Shannon information between the prior non-functional state to the posterior functional state, or vice versa, as I do in (4) above.
This is very idiosyncratic use of the term information gain and loss. I think this is why you are confused. However, as I agreed with you before, Delta H can be negative or positive.
The wikipedia page you reference does NOT define KL as the difference in entropy between to probability distributions. Why would you keep saying that when you can just look at the page and see that it is defined differently? It is defined, just as I explained before.
You can learn more about this from reading the page on KL divergence that uses the exact same definition in the wikipedia page you reference: Kullback–Leibler divergence - Wikipedia.
That calculation is in error.
KL is always positive. Information was required to move from a non cancerous to a cancerous state. You are making a very profound math error here.
The right way to think of this is different. The normal P53 state is well defined. We need to know how to change it into a carcinogenic state. There is a certain amount of bits required to specify how to change it into a carcinogenic state. That information is all functional information and it will always be positive because KL is always positive. If you used the formula that I just gave you, it would come up with a different number, a positive number.
Computing the precise amount of information in this case is another question. It turns out the mathematical framework you have can’t compute KL when the ground state isn’t IID MaxEnt. In this case, the ground state is normal P53, which is most certainly not IID MaxEnt, so that is why you are getting an aberrant result. You thought KL = delta H, but it does not. It never has except in the boundary case you stumbled into that has essentially no relevance to biology.
Notice the switch from p to q in the first term. KL looks a lot like a delta H, but it most definitively is not. The first summation in KL is notH ( p) or H ( q), because it includes both q and p. The second summation, however, is H(p).
Just to catch everyone up, if q is MaxEnt (the base state that @kirk is using), then in this case KL = delta H. If q is not MaxEnt, this is not the case. If q is MaxEnt, then for all p:
Here, N is the number of possible states. In this case it doesn’t matter that p is not q. Usually, however, it matters a great deal. If q is NOT MaxEnt, this not longer is true.
This a major change in your methodology. Here, you are using “normal” as ground state, rather than IID MaxEnt. That is much more justifiable and correct, however it is not what you did in your FSC paper. It also requires changing your formula. Using MaxEnt as a ground state, the carcinogenic p53 (by your FSC method) would be H(normal p53) - 4.2 bits, which would be hundreds of bits.
So, if you are going to propose using normal as the ground state, we should at least be clear that this is an admission that you are departuring from how you computed FSC in your paper. Failing to use an appropriate ground state wildly inflated FSC. We can go back and use the formula from your paper and we’ll see that it wildly overestimated FI for cancer, and therefore in the cases you applied it too.
1 Like
swamidass
(S. Joshua Swamidass)
Split this topic
36
@swamidass You nailed it. I totally missed that. Thank you for pointing that out. I see now that that definition is consistent with the other definitions of K_L; it is always positive. My apologies for the time wasted and I appreciate you pinpointing the problem.
I think we have made some progress here both by clearing up my mistake, as well as common agreement on (1) through (3) in my Post #7.
Just a minor correction here; since we are talking about only one site, it won’t be hundreds of bits. MaxEnt = 4.32 bits. Using MaxEnt as the ground state, we would get 4.32-4.2 = 0.12 bits, a gain in functional information for that particular site. However, that would be the incorrect way to measure the change in information from P53 in a healthy cell to P53 in a cancerous cell. The ground state for P53 in a healthy cell is not MaxEnt; it is the functional state.
This is not a major change in my methodology. I didn’t directly discuss loss of FI/FSC in my paper, but the groundwork is already there. Referring to my paper:
In my paper, Eqs. 2 and 6, establish the ground state as the general case. The null state is a special case of the more general ground state.
In my paper, the concept of functional information (FI) is generally defined by Eq. 6 as a deviation from the ground state, not the specialized null state.
On the ground state it reads, “The ground state g (an outcome of F ) of a system is the state of presumed highest uncertainty (not necessarily equally probable) permitted by the constraints of the physical system, when no specified biological function is required or present.”
It goes on to state that the null state is a special case of the more general ground state, and obtains when the physical system provides no constraints at all, resulting in all options being equi-probable. This means that the ground state must be determined by whatever the initial physical system is.
I then go on to estimate the functional information/FSC of a number of proteins by assuming the special case of the null state (i.e., no assumption that life already exists, nor translation, nor genetic code, and no a priori constraints on the probability distributions of amino acids in the universe). If one wants to separate out the FI provided by the genetic code, or allow for some other special ground state, then the reader would need to use the more general Eq. 6.
The more information we have about the ground state, the better. For example, in my current work I use the probability distribution determined by the genetic code as my ground state, not the null state. Obviously, this may be incorrect when it comes to proteins essential for building a ribosome translation system since the genetic code would not yet be in use.
The key issue is understanding the initial physical system well enough to accurately define the ground state for any problem, whether it is in biology, archeology, SETI, patent infringements, building combination locks, encryption, etc.
Back to the P53 example:
In the highly simplified, one-site example I provided in Post #7, we measured the change in FI to go from a healthy, functional p53 protein to a cancer-state, non-functional p53 protein. If, however, we want to start from scratch and code for a cancer-state, non-functional p53 protein, one way to estimate the FI for a non-functional p53 protein is to realize that a non-functional p53 has virtually no constraints imposed on H (ground). So H (non-functional) ≈ H (ground). Using my general Eq. 6, FI ≈ 0 bits.
In other words, it requires little or no FI to code for something that deviates little or not at all from the ground state.
I’ll be out of the office until Tuesday, so I look forward to picking up the discussion then.
Great. I’m glad we are on the same page on the definition of KL then.
Because KL is always positive, this forces several major changes to your understanding. You had thought that (in your toy case of P53), that cancer was an information loss. This is false. It requires a positive amount of information greater than zero, functional information, to move from the normal state to the new function of a cancer state.
That means cancer is gaining functional information. What remains is merely to compute how much.
I disagree. This has to do with the fact that you’ve only worked out the math assuming that KL = delta H. Now you realize this not the case, you have rework out the math. It turns out that the true amount of information is much more. This mathematical error we just caught has a massive ripple effect on all the calculations.
Yes, but this “incorrect way” is precisely how you measure the function of protein families. So behooves us to compute the information content of carcinogenic p53 in a way that precisely mirrors your paper until you are ready to concede your paper is in error. “Normal” p53 does not have the cancer function, but “carcinogenic” p53 does. In your paper, you would compute the FSC of the “carcinogenic” p53 in relation to a MaxEnt prior, ignoring the non-function “normal” p53. So that is what we should do here.
Unless you want to concede now that FSC is an erroneous way of computing FI. That is precisely where this is headed. The sooner we get there the better. I’m already there. Are you? Or do we need to play this out?
@Kirk, I’m looking forward to hearing your response to all this.
In general, I’ve been wanting to let you respond before putting another note. There have been a few loose ends along the way that are important to address though. You base your work on Hazen’s formulation:
You are deviating from Hazen’s formulation of FI. These deviations, in my view, invalidate your work. I wanted to point out a few of these here. For reference, this is the article that forms the basis of your work:
It is important to emphasize that functional information, unlike previous complexity measures, is based on a statistical property of an entire system of numerous agent configurations (e.g., sequences of letters, RNA oligonucleotides, or a collection of sand grains) with respect to a specific function. To quantify the functional information of any given configuration, we need to know both the degree of function of that specific configuration and the distribution of function for all possible configurations in the system. This distribution must be derived from the statistical properties of the system as a whole [as opposed, for example, to the statistical properties of populations evolving in a fitness landscape (37)]. Any analysis of the functional information of a specific functional sequence or object, therefore, requires a deep understanding of the system’s agents and their various interactions.
Therefore, to explore fully the distribution of function within a sequence space, a large number of randomly generated sequences (i.e., equal probability) must be surveyed (see Methods ). Such random explorations of genome space are similar to the strategies used in the directed evolution of RNA structures (e.g., refs. 47–48). Note, however, that this type of random sampling is not possible with living organisms because the portion of genome space explored in an evolution experiment will be constrained by the topology of the underlying fitness landscape and the particular configuration of the environment maxima (25, 49⇓–51).
Notice that Hazen, in this article, agrees with what I’ve said about this too.
Your response is that evolution can’t optimize proteins. I disagree. Your work certainly does not demonstrate this. Perhaps our best way forward is to agree that: “Your approach is invalid if it turns out that evolution can optimize functional proteins.”
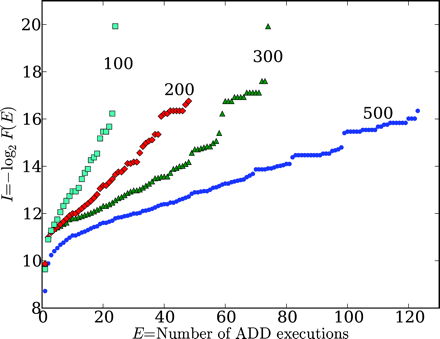
Longer Sequences More, not Less, Likely to be Functional
Next, Figure 2 is pretty interesting. They show that functional information decreases with increased sequence length in AVIDA proteins. This is opposite what would be expected by your formulation. I won’t into a long explanation here, but Hazen discusses this at length.
Without belaboring the point, you often quote Hazen’s work as support for yours. They we very careful to qualify the applicability of their work. It only works if sequences are uniformly sampled. Outside the context, such as in the case of extant sequence, the assumptions behind their work no longer apply.
I do not think it is appropriate to continue to cite is work given these oversights in your method.
Once again, this is precisely why cancer is a failure case that computes about 6 billion bits of FSC, or 6 billion bits of FI. Of course, this is erroneous, but that is what we have.
That is actually incorrect. None of my work was based on the Hazen paper, as a check of the dates on those papers will indicate. My paper was received on Jan 25, 2007 and Hazen’s paper was published some four months later in May 2007. I was not even aware of their paper when mine was submitted.
Neither my paper nor Hazen’s paper makes any reference to K-L divergence. Nothing in my paper depends upon it, therefore the fact that K-L divergence is always positive is irrelevant to my work. In the real world, we have cases of information gain and information loss. If a system of measurement is incapable of quantifying information loss, then it is of limited use when it comes to determining if information is lost or gained.
I am very familiar with Hazen’s work and find that, in general, their approach supports my own work. Even if he deviated from my work, it would not invalidate his work, nor vice versa. A major difference between the two papers is that the Hazen paper makes no attempt to estimate the functional information required to code for a protein, from real-world data. My paper is much more specialized in that it is primarily focused on describing a method to do it using real data. So he provides very little detail in that particular respect and more detail on other respects
Your recollection of my response is incorrect. What I have and continue to say is that the fitness gradients at the boundaries of functional sequence space are likely pretty steep (i.e., very selectable). You have not seen any of my work on this, so I’m unclear as to why you would believe that my work does not demonstrate this. The real-world data reveals that for most proteins (all the ones examined thus far), the belief that, within functional sequence space, there is a global maximum upon which the sequences converge, is falsified. The fitness gradients for certain species in a certain environment might be sufficiently steep to enable selection, but for the entire protein family, it appears there are a large number of local maxima and they are widely distributed in sequence space. Whether Hazen or you believe this or not is interesting, but says nothing about whether it is true or false. The data says it is false (and we have not yet looked at it, so I understand your reluctance to agree).
I am not sure I understand your response to my correction of your error when you thought that “hundreds” of bits were at stake for a single site in my toy example (and the toy example dealt only with a single site, not an entire p53 sequence). It should be clear that, at most, 4.32 bits would be at stake, but you said, “I disagree.” Are you saying that a single site, with only 20 symbols as options, can contain “hundreds” of bits?
It appears that you do not understand what I lay out in my paper. Referring back to Post #8, I actually showed how one would estimate the amount of functional information required to code for non-functional/cancerous cell P53, using the method in my paper. It turns out it is approximately 0 bits of information. It is at this point that we need to distinguish between how you would do it and how I would do it. It appears you are mistaken about what constitutes the ground state for a problem. We are not free to make up whatever ground state we wish; it is defined by the physical system, as I state in my paper. So let us take a careful, second look at the toy p53 problem I proposed.
Specified biological function to be examined: We wish to constrain p53 sequences such that it permits cancer (i.e., does not carry out it’s normal biological function of cell suicide).
Ground State: A healthy cell where the specified biological function to be examined, is absent.
“Functional” state: A cell that has become cancerous due to p53 failing to carry out its normal biological function.
As you can see, function is defined by the desired outcome. So if we refer back to my definition of ground state in my paper “The ground state g (an outcome of F) of a system is the state of presumed highest uncertainty (not necessarily equally probable) permitted by the constraints of the physical system, when no specified biological function is required or present”, it should be pretty clear in the toy example defined immediately above that the ground state is a healthy cell where the specified biological function to be examined is absent, and the “functional” state is as defined above.
To further drive this point home, Please refer to section (B) in my paper. Unfortunately, one of the required revisions of my paper was to radically reduce the number of mathematical equations and derivations, to be more in keeping with a biology journal. Consequently, all the math in section B was removed except for the very general Eq. 8. However, if you examine Eq. (8), and read that section, you will see that what I am proposing is precisely what I have done above. It should be very clear in section B that when it comes to measuring the change in functional information, we do NOT go back to a null state, or some arbitrary ground state … we must start with the physical system that we already have, and proceed from there … precisely as I have illustrated in the toy p53 problem.
Regarding the AVIDA model: Of course this is a computer model. As such, it is not in a position to falsify the real world data, which shows that larger proteins will require more functional information to encode. This should be patently obvious. Take 3D structure, for example, which is determined by physics. If a particular biological function requires a 3D structure that has three structural domains, we would not expect that the functional information to encode domain 3 would be less dense than what was required for domain 2. To use a different example, the number of symbols required to describe a microprocessor would be much, much greater than the number of symbols to specify the temperature of the room. We should not expect, however, that functional information required for the microprocessor is less dense than the temperature info. (unless the person describing the former is quite inefficient and unnecessarily verbose.
The issue to be resolved: We need to come to a clear understanding of what constitutes the ground state, for any problem. Even if, in the end, you do not want to agree, I can show for a wide variety of real world problems that my method will work and identify both gain and loss of functional information.
It is the same research as Sostack and it is often quoted alongside your work. And I agree with their assessment. In fact, your formulas exactly matches theirs. Though it turns you DO build it on their aprpoach. Here are a list of references your made:
Maybe, but that is because you thought delta H was the right quantity to compute. It is not, unless you are uniformly sampling sequences. However, in your case, you are not.
You reference Hazen more than anyone else. You work certainly is related to his. This specific paper was submitted afterwards, but I’m not sure what your point is. They have published on this a bit, and have correctly caveated their work in ways. Your work violates the assumptions required to compute FI.
I am echoing the limitations of their work that your work ignores.
I’m not really interested in getting into a dense textual argument with you. The math is a lot more clear, at least to me.
Yes, I know that you disagree. It is just good to know that the entire computation depends on agreeing with you here, and I do not, nor do most biologists. It is just chalked up as an assumption that you think you can support and with which we disagree. I’m fine with that for now. No reason to belabor the point.
No Reference to KL
Really @Kirk? You seemed to be pretty clear that you were wanting to compute KL just a week or so ago. What happened? What changed?
So if we are not computing KL, MI, CD, or Joint Information, what are we computing? Delta H? Why is delta H the right quantity to compute?
I can show you why KL is the right quantity to compute. KL divergence gives us the number of bits required to tell us how to move between one state and another. In the case of p53, it is the number of bits to move from the normal state to the cancerous state. In the simplified example here, where we must mutate a specific amino acid to something else, It is not 4.2 bits. It is about 30 bits.
Delta H does not give us the amount of information required to move between two states. So there is no reason to use it. Why have you decided that delta H is better than KL distance? Now that we have established they are different things, why would we want to compute a different number than the number of bits reuqired to specify how to move between two states?
Yes, because this mathematical framework cannot apply to real world data as you have done it.
P53
It is not hundreds of bits. It is about 30 bits. In cancer, we need several of these sorts of changes, so that soon becomes greater than 100 bits. Do you see why it is about 30 bits?
This is a deviation from the method you used in your published paper. It is irrelevant to our conversation. What we should be computing the problem using this:
Ground State:Uniformly random sequences of the same length as p53.
That is the ground state you use when measuring FI from pfam families, so that is the ground state we should use here. Right?
Notice also that you are claiming that the information gain is 4.2 bits or 0 bits (as the case maybe). That means that something like 1/16th of all proteins are a carcinogenic p53, using the formula FI = -log(M(Ex)/N). That doesn’t make sense. So clearly your framework is breaking down. The reason why is because you can’t use delta H this way. Also the FI formula you are using breaks down if you aren’t sampling uniformly. If you use normal p53 as your base state, neither of those formulas apply (neither delta H nor FI = -log(M(Ex)/N)), and you have to use KL divergence instead.
Regardless, your appraoch to measuring the FI using pfam uses a maxent groundstate. So that is what we should use here. That is how we understand how using the maxent affects your results. Unless of course:
This is a major point @Kirk. When you computed the FSC of a protein function, you udse a maxent ground state. In this case we should too, unless you are willing to throw out all the computations you’ve made using a maxent ground state. If you are willing to do that. Great. Just say so. If not, I insist that we use the same ground state as your work does.
We agree that the carcinogenic p53 is a well defined function. We agree that we can apply your approach to it. So let’s apply your approach.