I wrote a simple script that takes a list of 100 sample accession IDs and downloads the corresponding sequence data. Based on the BioProject metadata, I believe these sample are from the New Mexico Department of Health Scientific Laboratory and were collected in August of 2020.
Again, these samples were chosen entirely opportunistically and are likely not representative of anything in particular. Obviously, this is would not be a good study design if our goal was to say something useful about genetic variation in SARS-CoV2, but it should be okay okay for a simple tutorial.
Each sample is represented by two files. For example, accession SRR12638318 consists of these two files:
SRR12638318_1.fastq.gz SRR12638318_2.fastq.gz
These data were produced via so-called “paired-end” sequencing. In this process, genomic DNA is sheared or fragmented into a range of sizes (typically a few hundred nucleotides, although this will vary depending on the particular protocol used). And then each end of the fragmented is sequenced, generally starting from the outside and moving inward. Here is an illustration from Illumina, the company who currently has the market cornered on this particular technology:
So for each DNA sequence fragment, we will have two “reads” – one (depicted as orange above) sequenced in the “forward” direction, and another (blue) sequence in the “reverse” direction. All of the forward reads go in one file, and all of the reverse reads go in another. Hence, two files per sample.
These data are in the “fastq” format, which is somewhat similar to “fasta” but a little more complicated. Here is what the data for a single read look like:
@SRR12638318.2 2 length=151
TGCCTGTACCAAGAAATTATGATTAGACTTACGAATGAGTAAATCTTCATAATTAGGGTTAAGCATGTCTTCAGAGGTGCAGATCACATGTCTTGGACAGTAAACTACGTCATCAAGCCAAAGACCGTTAAGTGTAGTTGTACCACAAGTT
+SRR12638318.2 2 length=151
11>>A1F3DFBFGFCBFG1GBBGHBGBGGHFHCACGBFHHGFGHHHHHFHAFGHHGHHGHHHHHHHHHHHHHHHHHGHHHHHGHHHHHHHHHHHHHHHHGHHHHHHHHGHHGHHHGGHGHHHHHHHGGGGGHHHHHHHHHHHHHHHHEHHH
The first line is the sequence identifier and starts with an “@”. Typically this line will provide information regarding the sequencing instrument, the position of the read in the flow cell, and whether the read is forward or reverse (among other things). But in this case the data have been downloaded from the SRA repository, and much of this information has been removed, leaving us with just the accession ID and the read length.
The second line contains the actual sequence for the read. Pretty straightforward.
The third line is mostly just a placeholder to separate the sequence line from the quality scores line. It always starts with a “+” and may optionally be followed by other information (a repeater of the identifier line in this case).
The fourth and final line consists of quality scores for each nucleotide in ASCII format. Basically, the sequencing machine attempts to estimate the probability that the base was incorrectly called. In the above, an “H” represents a PHRED quality score of 39, which is a 0.00013 chance of an error. That’s a very good score, although there are various sources of error that are not accounted for in PHRED scores (see here for more info).
That’s the fastq format in a nutshell. For each sequence, there are 8 total lines spread across two files (forward and reverse). Because of this, the data add up very quickly, and the files can end up taking up a lot of space.
In this case, though, we don’t have too many reads per sample. As we’ll see later, it simply doesn’t take all that much sequencing to thoroughly cover a 30,000 nucleotide genome!
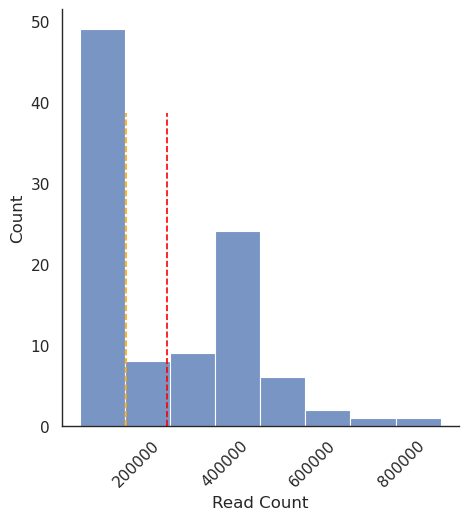
Here are the mean and median read counts:
Average reads per sample: 271432.66
Median reads per sample: 176628.0
And here is a histogram of the read count data, with mean and median highlighted by red and orange dashed vertical lines:
Now let’s look at the length of the reads:
Average read length: 117.49
Median read length: 136.0

I believe each forward and reverse read were originally ~150 base pairs in length (that’s the most common read length typically used in paired-end sequencing these days), but it appears that these reads have gone through some quality control that has altered the length distribution. Typically, the PHRED quality of the read decreases as the length of the sequenced molecule increases. Most QC tools will trim a read if the average length falls below some threshold. Therefore after QC, you get a large peak in the length distribution that is close to the original read length, followed by a tail of reads that have been trimmed to various lengths in order to remove low quality sequence data. That appears to be what happened here.
Okay, that was long and fairly boring, but hopefully the nature of the data we’re using is a little bit more clear. I’m going to leave it here for now and pick it back up tomorrow or in a couple days. The next step will be to align the reads to the reference genome.
In the meantime, feel free to let me know if there are any questions regarding things I didn’t cover or didn’t sufficiently clarify. I’m happy to correct any errors, so please point them out if you see any.