My claim is that there is no evidence in the wild that sarbecoviruses can recombine with other subgenera of betacoronaviruses. Here are some quotes from the paper I cited that support this claim:
In the Abstract: We find that recombination accounts for nearly 40% of the polymorphisms circulating in populations and that gene exchange occurs almost exclusively among strains belonging to the same subgenus
In the Author summary: The Betacoronaviruses comprise several populations that could be considered distinct biological species in that they do not engage in gene flow with one another.
In the results section: To distinguish between these alternatives, we simulated the mutational process—without recombination—by adding similarly diverged sequences to the set of genomes in a focal subgenus. Under these conditions, addition of a sequence simulated exclusively with mutations to each focal subgenus recapitulated the observed pattern of h/m reduction (S3 Fig), indicating that none of the three focal subgenera recombines with one another.
In the discussion: By analyzing the diversity within natural populations of Betacoronaviruses, we find thateach of the major subgenera–the Embevoviruses the Merbecoviruses and the Sarbecoviruses–is reproductively isolated, with gene exchange restricted to members of the same subgroup.
Now, I realize that you’ve cited some other passages from the results section that appear at first sight to contradict the above quotes. What can be said from this? As for me, I don’t really know.
This quote, taken from the Methods section of the article I referred to in the post below:
#### Recombination analysis
Within the framework of ConSpeciFix , we evaluated whether polymorphic sites within and among clades of Betacoronaviruses were more likely to be attributable to mutation or to recombination. We used the three largest subgenera, Embevovirus ( n = 81), Merbecovirus ( n = 17) and Sarbecovirus ( n = 45), as focal clades, and used a resampling approach to estimate the ratio of homoplastic ( h ) polymorphisms (sites that could not be parsimoniously ascribed to vertical ancestry; i . e ., recombinant alleles) to non-homoplastic ( m ) polymorphisms (vertically transmitted alleles) for each subgroup.
So it seems that the sarbecovirus subgenus comprises more than three viruses.
You want to move the goal posts from “SARS2” to all “sarbecoviruses”, but that won’t work and that’s why my response focused solely on detecting recombination between SARS-CoV-2 and other subgenera of betacoronaviruses which are not phylogenetically closest to it.
As per the paper, recombination between the various subgenera is extremely rare and that’s something I never denied. Of course, something being rare doesn’t mean it can’t happen and we saw just that for SARS-CoV-2.
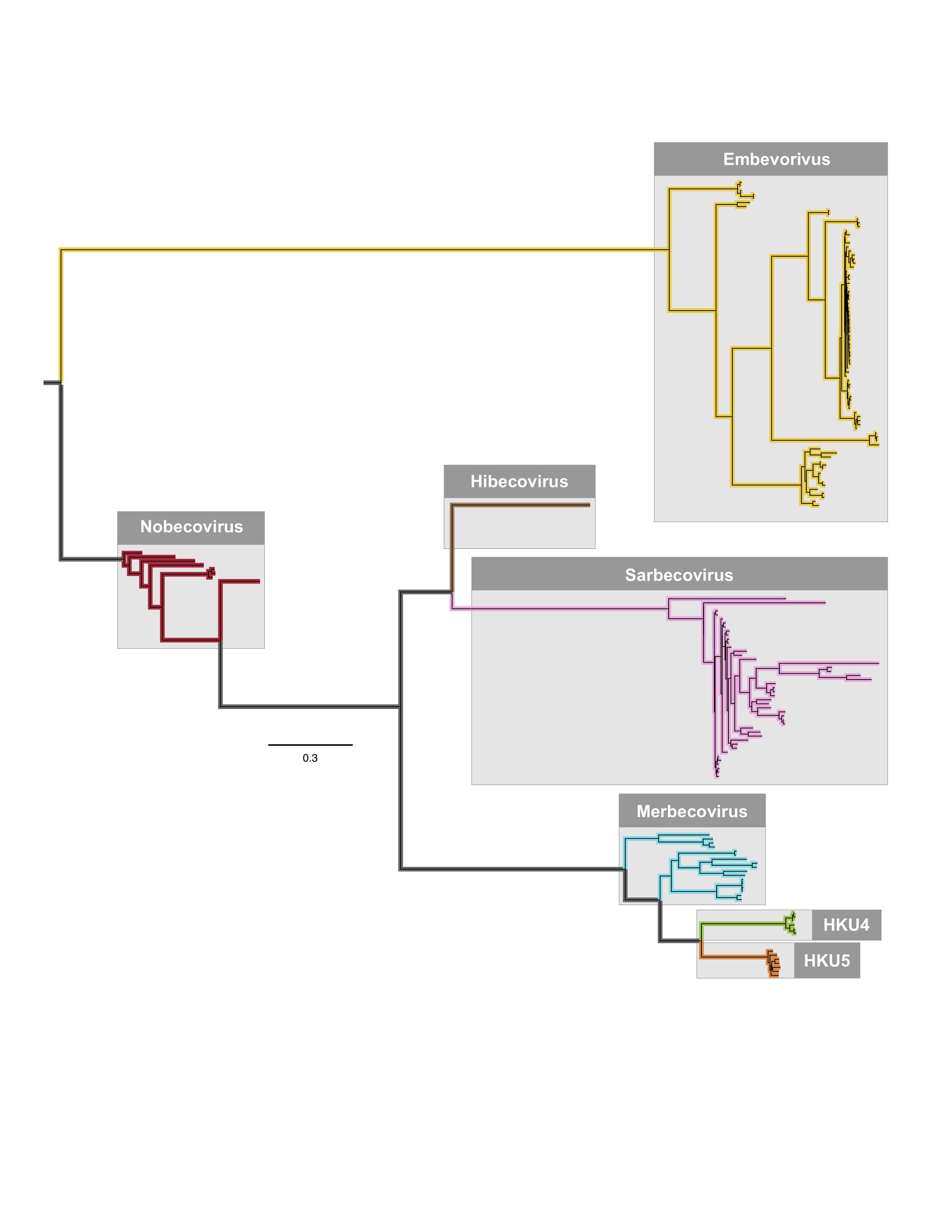
However, it seems as the phylogenetic distance closes between the various subgenera, recombination becomes more likely: merbecoviruses are more closely related to sarbecoviruses, than sarbecoviruses are to embevoviruses and this could explain why recombination between SARS-CoV-2 was seen in merbecoviruses and none (based on their small dataset) with embevoviruses. This is the betacoronavirus phylogeny from the paper:
This is an important omission you made because my previous response has been to demonstrate evidence of recombination in the genome of SARS-CoV-2 and other distantly-related betacoronaviruses. Reread my response and see that. Remember you claimed that only those strains closest to SARS-CoV-2 could have recombined with it, but the paper you cited contradicts that claim because it provided evidence for earlier recombination between SARS-CoV-2 and more distantly related betacoronaviruses.
Did I deny this?
You are also forgetting that this (gene flow) is not impossible, but extremely unlikely as the authors note in their abstract. More importantly, this does not hold for SARS-CoV-2 because the data says so and the authors acknowledge this. Look at the excerpts again:
And:
There, my friend, is the evidence for recombination between SARS-CoV-2 (a sarbecovirus) and merbecoviruses (which are phylogenetically distant). As a bonus, they indicated some of these detected recombination was between SARS-CoV-2 and merbecoviruses in Wuhan, China.
Your claim is defeated. Period.
Yes this seems to be the general trend but there are exceptions and SARS-CoV-2 is one of them.
What can be said from this is that you have ignored the clear evidence of recombination between SARS-CoV-2 and other subgenera of betacoronaviruses in the very study you cited.
In addition, I am also curious why you accept virus phylogenies but not that of organisms? (@BrianLopez, do you see another example of an ID proponent accepting phylogenetic evidence in one context but ignoring it in another)
The evidence for recombination between sars-cov-2 and merbecoviruses is not as compelling as you seem to think for it leaves room for another less likely but still plausible possibility.
Thanks, looking over the nucleotide alignments they produce in that paper I’m not persuaded they represent a more likely scenario than the alternative they propose.
Also, I honestly don’t follow their assertion that the scenario they propose is more parsimonious:
A conclusive proof of any novel insertion is the existence of closely related strains without it. In the case of SARS-CoV-2, the PRRA insertion is obvious because closely related strains RaTG13 or Pangolin/GD/2019 do not have the PRRA fragment while still having the nearly identical nucleotides around the same locus where SARS-CoV-2 has the insertion. In the case of RmYN02/RacCSxxx, the purported PAA/PVA insertion is always coupled with a purported 4 amino acid deletion just preceding the NSPAA/NSPVA fragment. This deletion corresponds to a QTQT fragment in SARS-CoV-2, RaTG13 and Pangolin/GD/2019. If PAA/PVA truly was an insertion, one would expect to see closely related strains that do not yet have that insertion but already have the purported 4 amino acid deletion. In the absence of such strains, the more parsimonious explanation for the PAA/PVA fragments is not a 3-aa insertion combined with a 4-aa deletion, but point mutations and a 1-aa deletion instead.
In a locus already heavily prone to recombination, a deletion and an insertion seems to me just as parsimonious(actually more parsimonous, as it involves fewer total events) as a deletion and three or more substitutions creating a FCS.
But that does raise another issue, which is that there really is a naturally evolved FCS in RmYN02, another sarbecovirus, it’s just that (they argue) instead of originating by insertion, it apparently evolved by a combination of a deletion and a handful of substitutions. So that FCS is now a candidate for recombination among it’s related sarbecoviruses. It seems to me entirely plausible that there is still lots of unsampled betacoronavirus diversity out there, some carrying FCSs that could be the source of SARS-Cov2’s.
In what way is SARS-Cov2 an exception to the general trend? The origin of SARS-Cov2 is the case we are trying to assess, so it would be begging the question to simply invoke it as a virus who’s FCS originated by recombination from a more distantly related betacoronavirus.
Oh its quite simple. Gill is not convinced by the phylogenetic evidence that humans and chimps shared a common ancestor, but he is convinced that SARS-CoV-2 and RATG13 shared a common ancestor, despite the fact that the phylogenetic evidence in both cases are essentially derived the same way. This shows he accepts phylogenetic evidence only when it lends some degree of support to his beliefs (in this case, a lab leak).
Are we looking at the same data? If we are, then you should know this is a ridiculous statement. The h/m ratio for recombination between SARS-CoV-2 with other sarbecoviruses and merbecoviruses in the study was essentially the same. Look at the data in case you have forgotten:
According to the paper, genetic exchange between strains of the various subgenera is quite rare and rare enough to consider them as biological distinct species. This means we shouldn’t detect recombination between SARS-CoV-2 and other betacoronavirus subgenera. However, it turns out there was significant recombination between SARS-CoV-2 and merbecoviruses (but not embevoviruses). This makes SARS-CoV-2 an exception to the general observation that strains from one subgenus of betacoronaviruses does not recombine with strains in other subgenera.
I am not arguing that the FCS of SARS-COV-2 derived from recombination with some other distantly related betacoronavirus (although this would be the case for several segments of its NSP genes). I was rebutting Gil’s claim that only phylogenetically close strains could have recombined with SARS-CoV-2 and as the paper he cited showed, that is clearly false (but he didn’t realize this and went ahead to cite it).
With regards to the origin of its FCS, we should look more to recombination with other sarbecoviruses as that is the most frequent. As the paper showed, there is plentiful recombination between SARS-CoV-2 and other sarbecoviruses in the spike protein region, so it’s quite likely it got its FCS (or a pre-FCS which would go on to mutate to the current form) via recombination with another unknown sarbecovirus.
Although this observation favours the idea that SARS2 may have recombined with merbecoviruses, it doesn’t exclude the possibility that it could have resulted from convergent evolution, whose probability of occurrence, according to the authors, is 5% (see below).
Of particular interest, we detected an array of 11 clustered homoplasies with similar genome distributions in SARS-CoV-2 and two genomes of bat Merbecoviruses isolated from Wuhan China (MG021452 and MG021451). However, simulations revealed that this pattern has ~5% probability of resulting from convergent mutations given the extent of saturation in the dataset
So I stand to my claim you think is ridiculous, ie., evidence for recombination between sars-cov-2 and merbecoviruses is not as compelling as you seem to think for it leaves room for another less likely but still plausible possibility.
Seriously? Quit embarrassing yourself please. Its now even more ridiculous to ignore the ~95 probability that the observed pattern was due to recombination and not convergence. If ~95% isn’t more compelling relative to ~5% to you, it seems you need a reality check. More so, the authors indicated this 5% probability of convergence (~95% probability of recombination) for just two bat merbecovirus genomes and SARS-CoV-2, because the merbecovirus sequences were collected from Wuhan, China: its not for all other cases of recombination between SARS-CoV-2 and merbecoviruses.
Remember the abstract wherein the authors admitted that gene flow between the various subgenera is possible, but extremely rare, it is data like the recombination observed between SARS-CoV-2 and merbecoviruses which prompted that admission. The authors are not on your side, and so is the evidence (more importantly).
I think you are also ignoring another key finding in that paper and that’s the high frequency of recombination between SARS-CoV-2 and other sarbecoviruses in the spike protein region where the FCS is found. That makes recombination (and probably subsequent mutations) a very likely cause for the presence of a FCS in SARS-CoV-2.
You are the one that need a reality check, for I’ve explicitly stated that the recombination hypothesis is more likely (and so more compelling) than the convergence one. For the record, here is what I said: evidence for recombination between sars-cov-2 and merbecoviruses is not as compelling as you seem to think for it leaves room for another less likely but still plausible possibility.
Come on Gil, we can all read your comments. That is clearly not what you said. You stated:
You thought the evidence for recombination was not compelling due to the ~5% chance that convergent evolution accounted for the patterns of homoplastic clusters observed in the genomes of SARS-CoV-2 and the two merbecoviruses. You just ignored that the probability of recombination accounting for that pattern was ~95% which is very compelling and why you needed a reality check.
In any case, you now seem to have agreed that the evidence for recombination between SARS-CoV-2 and merbecoviruses is “more compelling” (probably after realizing your error) based on the data. That’s a common ground we now share.
Furthermore, the spike protein seems to be the most important factor that allowed SARS-CoV-2 slip into human populations and as per the data in the paper you cited, recombination between the sampled SARS-CoV-2 and merbecovirus genomes took place outside the spike protein region, which probably makes them less important to investigations on the origin of SARS-CoV-2. However, the rampant recombination between sarbecoviruses in the spike protein region is a strong indicator that the FCS probably derived from it. This observation (rampant recombination in the genome sequence of the spike protein) and others are what make the zoonosis hypothesis remain the most plausible explanation for the emergence of SARS-COV-2.
That makes it the most plausible explanation for the origin of the SARS-CoV-2 spike gene. It doesn’t really tell us about how the virus got into humans.
That makes it the most plausible explanation for the origin of the SARS-CoV-2 spike gene. It doesn’t really tell us about how the virus got into humans.
Steve did you by any chance miss the part in bold:
I was looking at other pieces of data as well. Nothing unsettles your comment though, because recombination is the most likely explanation for just the natural origin of the 12 nt insertion in the FCS of SARS-COV-2.