Hi Faizal
The challenge is not in the number of genes as different genes are assumed equally probable getting the mutation.
The assumption that all amino acids are equally substitutable (as in your example) is not consistent with the evidence.
As you add to the length of the sequence the waiting time gets exponentially longer. The exponent depends on how substitutable the amino acids are. The assumption in the Behe/Lynch case is that 6 work in every position.
What we are testing is if the waiting times are realistic given the mechanism we are proposing. If animals were seeded on earth de novo there is no waiting time issue. What you have proposed is untestable.
And the only way they could be fail to be “realistic” is if we begin with an axiom from which it is entailed that they could not. Since this axiom is without any basis, and is actually contradicted both by experiment and historical inference, there is actually no waiting time problem.
You don’t need to wait for this because it happens all the time. A single protein can bind a diverse number of ligands and a single ligand can be adsorbed to different proteins with varying levels of ease. For example, if a protein’s binding site has amino acid residues with charged side chains, then it can interact favorably with ligands containing (oppositely) charged groups even if those interactions are weak or uneventful. Proteins bind different things all the time; some of these interactions will be beneficial and may be kept by natural selection; some others might be deleterious and get purged by negative selection; some others might provide functions not useful or harmful to the cell and others will simply be uneventful.
No response is possible to your comment, as barely a word of it is coherent.
A suggestion: Quit trying to impress people by writing like how you imagine a scientist would write, and just communicate your thoughts in the English you usually use.
Hi Faizal
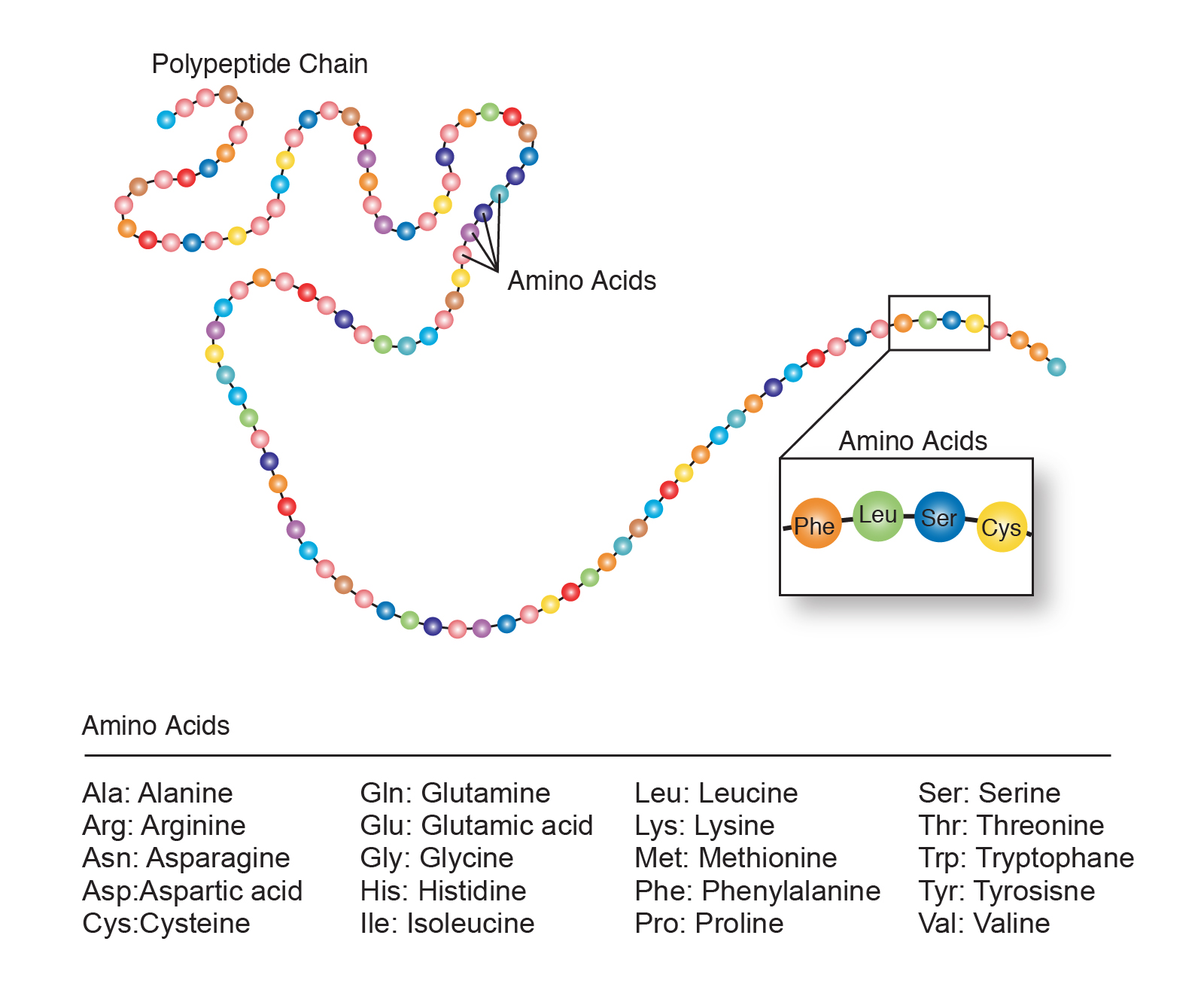
Each protein position has one of 20 amino acids. When I say substitutable it means how many of those 20 will work in each position. Below is an example of a protein string prior to folding. At the bottom are a list of the 20 amino acids.
Since proteins are sequences of amino acids the chance of a mutation working is dependent on how many amino acids will work in every position. The problem gets bigger (exponentially) as the length increases. Assuming half work in every position every time you add an amino acid the amount of non functional sequences doubles.
The counter argument is that almost all work. The problem with this argument is that we observe functional constraint in biology.
We also need differential binding meaning not all proteins can bind with each other or cellular functions would not work properly.
In the Behe Lynch discussion the assumption is that 6 amino acids out of 20 would work in every position.
No, that isn’t a counter argument anyone has made.
The counter argument is to explain your continued mistaken focus on particular structure-function relationships, which commit the Texas sharpshooter fallacy. That is, you are focusing on a particular sequence adopting a particular structure to perform a particular function, instead of understanding that evolution just needs to find some sequence performing some adaptive function.
Besides that, there’s the fact that only evolution actually makes sense of the distribution of novel genes, and particularly their characteristics. Bill what is your response to this post?:
If, at every site, there is a 50% chance that a mutation will result in a functional protein, then there is a 50% that any mutation will result in a functional protein for the protein as a whole. It does not matter how long the protein is.
And if a protein is longer, that does not necessarily mean that more mutations are required to change it to one that performs a new function.
The waiting time is the process of “getting there”. In this case it is time to wait for a successful gene duplication and the time based on mutation rate and the number of substitutions required to “get there”
Waiting time is one of the parameters in the Behe Lynch discussion along with required population sizes.
Broadly speaking, the waiting time is the time difference between not having something and eventually having that thing. It is not a process. You just love to say contradictory stuff.
Guess what Bill, evolution doesn’t want to “get there”, it “goes anywhere”. If one, two, or ten mutations (be they duplications, insertions, substitutions, deletions, etcetera) suffices to get some adaptive function, then evolution will work with it and eventually optimize that function with future mutations.
Wrong. That only applies to Behe’s model, which is a variant of the Texas Sharpshooter fallacy.
Lynch’s model includes the variable n, which is the number of possible sites that are available to mutate and produce a functional protein. So, for instance, a new protein might require 2 mutations, but if n=10, then these mutations could occur at any of 10 loci. Behe did not account for this, which is one reason his results were unrealistic.
And it should be obvious that the larger the protein, the larger n is likely to be. So a larger protein is no less likely than a smaller one to producer a new functional protein,
Just a note: The expected median waiting time for an event that occurs at a rate of 1/N is approximately 0.7*N ( in the units of N). The expected waiting time will be considerably shorter if the is more than one possible sequence that can perform the same function sufficiently. The waiting time will be zero if such a sequence already exists somewhere in the genome, and the new function is activated by the initial duplication/mutation.
The probability of pre-existing mutation will depend on how much of the genome is available to accumulate mutations, or may already have a similar function that can be exapted. I don’t know how to estimate that, but it’s got to be more than zero.
This is a good frame for reminding @colewd that he’s still understating the numbers of human variants of that MYH7 gene encoding an incredibly complex protein by several fold.
What condition makes the waiting time realistic across a broad group of proteins? If neutral mutations were expected we would see a lot of sequence divergence between rats and mice given the time from their theoretical split and reproduction rates. What we see instead in several cases is much less divergence then neutral theory would predict.
Given this I don’t think Lynch’s model applies to vertebrates. A gene involved in development, beta catenin, we would expect a divergence of greater then 30% under neutral theory and the actual divergence is less then 1%.
Hi John
Based on a uniprot alignment the divergence between Rats and Mice on the MYH7 protein is less then 1%. If we add humans to the mix and compare all three sequences it is less then 3%. This does not fit the model that MYH7 can mutate neutrally in these populations.
Bill, you’re only comparing CONSENSUS sequences. You’re just pretending that a massive amount of directly observed human variation doesn’t even exist. Why?