Are you spying on me while I write? There is no way you had time to read and write since I posted. I know that a lot of the chemical interactions are similar. I know that promiscuous enzymes can carry out multiple reactions of different types. I know they can be pushed to favor one over the other. But not many enzymes are as promiscuous as cytochrome P450, whose function is to break down all kinds of gnarly compounds.
And yes, BRENDA does categorize by the kinds of transformations that take place. But these reaction categories have meaning. They can’t be squished into one thing. Otherwise David Baker’s group wouldn’t have to work so hard to pick out the right fold for his new chemistry, or Francis Arnold would not need to develop elaborate mutation and selection schemes to get the shifts in function she and her group do.
You’re going off the rails. Is ADP the substrate or the product of myosin? What is the chemical reaction catalyzed by the enzymatic part of myosin?
And beyond that, you keep missing the complexity of myosin here, which is why I gave this a low chance of working, much less working beautifully with two different myosins. It’s not a simple enzyme. It’s a motor. It astounded me that we could take such a crude approach and obtain such elegant results. We could take the approach that worked for far simpler kinases and apply it to myosins because function of all kinds is easy to find in sequence space.
You are gravely mistaken.
How does rigor mortis work again? What happens if I mix wild-type myosin, actin, and ADP together?
No, that function never existed. You are mistaken.
Correct. Now, what does wild-type myosin do in the presence of regular ADP?
That is false, Dr. Gauger. Binding actin is a critical part of the myosin cycle, which is covered pretty universally in undergraduate biochemistry IIRC. How can myosin move relative to actin without binding it?
Maybe you should rethink that claim, because you are confusing substrates and products.
I think I’ve been very clear that I’m not looking for the evolution of a new trait. This was engineering to help us to figure out what these myosins do. I am pointing out that function is easy to find in sequence space, even bifunctionality, because the mutant myosin is perfectly functional.
I don’t think there’s any point to this if you’re going to deny things covered in undergrad biochemistry.
No myosin moves actin in the presence of ADP, so the mutant’s binding to actin in the presence of the ADP analog is perfectly functional.
When we do in vitro motility assays like this one:
in which we have myosin bound to a glass coverslip moving fluorescent actin filaments, we have to include an ATP regeneration system to immediately remove the ADP produced by myosin’s hydrolysis of ADP. Otherwise, the ADP halts the motility.
I concur. BREDNA is a useful tool for some questions but there remains significant ‘fuzziness’ in applicability when we want to consider protein evolution. It can be a case of mistaking the forest for the trees.
It would seem to me that mutations in the beta-lactamase gene could result in an enzyme that breaks down a different substrate. If you only test for beta-lactamase activity you will miss this change in function. In my experience, enzymes are rarely so specific as to only cleave one very specific substrate. I once tested a protease against an array of thousands of substrates and found hundreds of hits for the protease.
What I balk at is the very limited view that a protein can only ever have one function. Therefore, all you have to do is test against that one substrate. If you want to make grand claims about general function then you need to look at more than one substrate. I also balk at the idea that any peptide can be claimed to have no function given the millions and perhaps billions of biologically relevant substrates and protein-protein interactions.
I stand corrected. ATP is the substrate, ADP the product. But both are intimately involved in myosin’s action. If ADP does not leave its binding pocket actin and myosin stay frozen together, and the cycle of myosin’s movement along actin cannot continue. You say
Is there some equivocation here? Both parts of your sentence are technically correct, but I said
You said
The abstract of your paper said
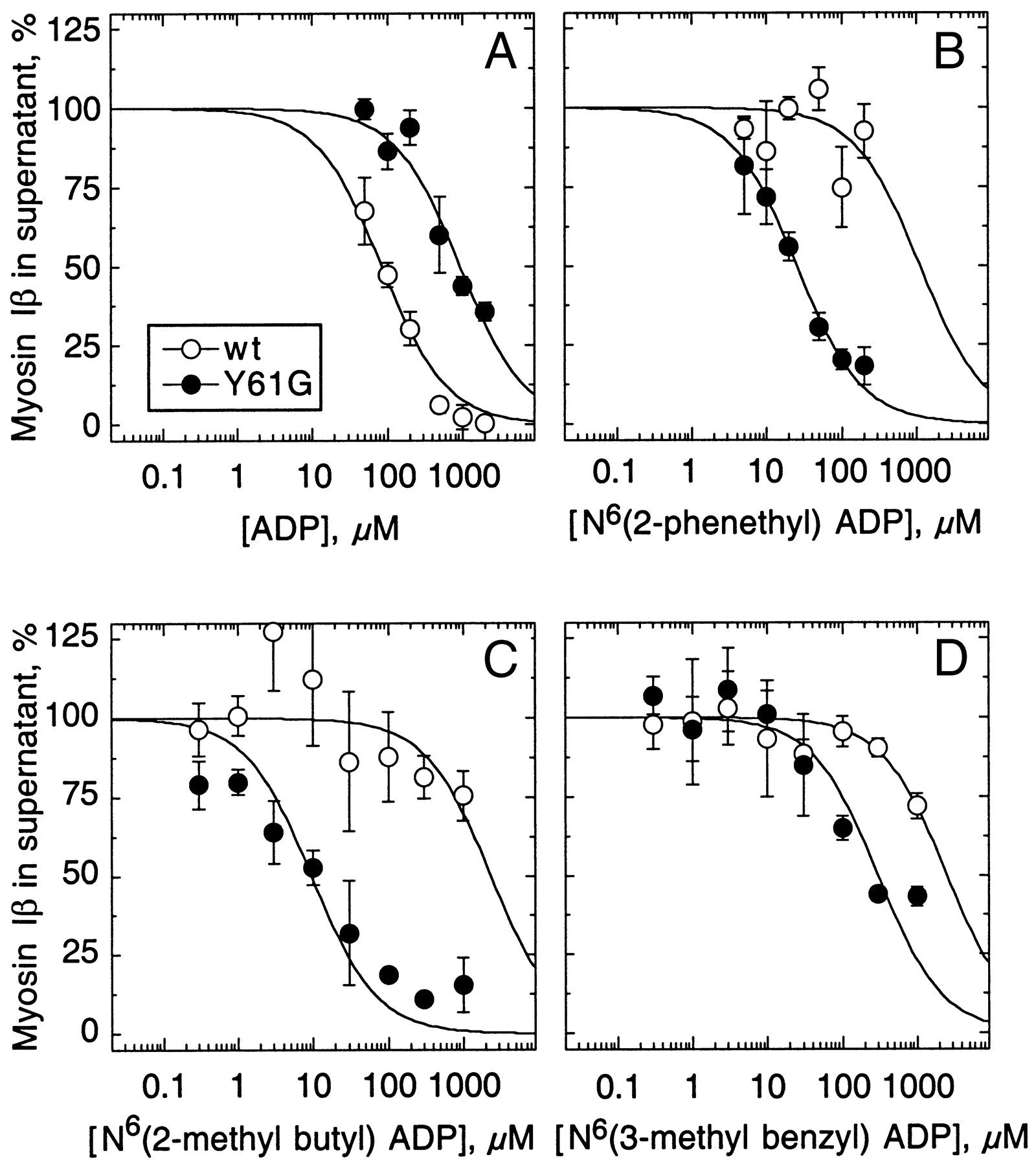
in doing so, these analogs locked Y61G myosin-Ib tightly to actin. N 6-(2-methylbutyl) ADP abolished actin filament motility mediated by Y61G,
Y61G is the mutant myosin, and N 6-(2-methylbutyl) ADP is the analog ADP. If the analog does not dissociate, myosin cannot release and movement ceases. Yes, the analog binds, but too tightly.
OK< let me just talk this out. Normally myosin binds ATP, hydrolyzes it, and the ADP plus inorganic phosphate remain associated with the myosin in high energy state, cocked. The myosin binds actin, the inorganic phosphate releases, and myosin executes the power stroke. Normally the ADP releases. In a normal cell ADP is quickly cycled to ATP. Now let me ask you this. With analog ADP, does it release from myosin at all? My guess is no, because you use this mutant myosin and the analog to selectively freeze certain kinds of myosin.
No, both the ADP and the ADP analog are cycling. Our use of these terms is in the context of ensembles of myosins. IOW, only an ensemble of myosin heads can move actin in the in vitro motility assay.
Yes. It’s not in the paper, but the mutant hydrolyzes the ATP analog just fine (those experiments are far more expensive than ones with the ADP analog).
@Mercer Then why does the abstract say that the mutant binds actin tightly in the presence of the analog, and cannot move actin? Is the power stroke missing?
Is your scheme to put the mutant myosin in to replace one isozyme of myosin, then add excess analog to the cells somehow. The analog is only bound by the mutant myosin, thus putting the actin/ mutant myosin into “rigor” but leaving other myosins unchanged.
So if your mutant myosin can bind and hydrolyze analog ATP, and can bind actin, does it complete the power stroke with the analog bound? It will with ADP. What about analog ADP? I realize I am asking a lot of questions that are probably found in your paper, but getting this right is important.
Why does the mutant myosin fail to move myosin in the presence of the analog? Does it fail to assemble into an ensemble?
The abstract is saying that because wild-type Myo1c binds actin tightly in the presence of ADP. The point is that the mutant behaves normally.
May I respectfully suggest that you read beyond the abstract?
Abstracts are intelligently designed to help the reader know if the paper they precede is of interest, not to provide all the information from the many experiments in the paper.
Yes.
We also have 3 negative controls for specificity: cells without the mutant myosin but with the ADP analog (always provided with a lot of ATP), cells without the mutant myosin and without the ADP analog (but provided with the extra ATP introduced with the analog), and cells with the mutant myosin but without the ADP analog (but with ATP).
One thing you’re missing is that we don’t have to replace the wild-type version, although we have done so. Since these myosins work as ensembles, even a small minority of myosins in the rigor state is enough. Our first experiments were done with inner-ear hair cells from transgenic mice, with lines chosen for their low copy number and expression levels.
I’m sure that it does based on the biochemistry, but don’t think it would be worth the time/money to do those experiments (no reviewer, through 2 grant applications and 5 papers, has asked for them).
In the presence of the ADP analog, it binds actin strongly, just like the wild-type myosin fails to move myosin in the presence of regular ADP. That’s why we have to clean up any ADP produced by hydrolysis and convert it to ATP when doing in vitro motility.
Most proteins interact with several substrates, but the range of kind of substrate is not broad for most. I suspect enzymes with internal binding sites or complex multistep reactions are more constrained. However, their specificity can be broadened by enlarging their active sites.
Beta lactamase is a hydrolase. Its range of antibiotics it targets can be enlarged by enlarging the active site. But they are all members of the beta lactam class of antibiotics.
It would be possible to ask if beta-lactamase can replace any function in E coli, for example, or some other disease-causing bacterial strain, but i suspect it will substitute for few if any other enzymes.
A relevant study was done by Patrick et al. They identified 120 enzymes required for biosynthesis of amino acids, purines and pyrimidines in E coli, and then obtained stocks deleted for these genes. They then transformed each strain with a plasmid library containing over 4000 E coli genes, and looked to see if any gene could rescue the deletion mutant and allow it to grow on minimal media (lacking amino acids, purines and pyrimidines). Normally E coli is able to make all these compounds, so if the missing gene can be substituted for somehow, it tells us there is a candidate for promiscuous rescue somewhere in the Ecoli genome. Self rescue was a control for completeness of the transformation.
They found about 20 deletion mutants could be rescued. Some of the rescuers were isozymes, some were metabolic work arounds, and about 6 looked like they could be related enzymes capable of substituting. A few were unknown. 20 rescues seems good, but remember over 80 could not be rescued. And rescue occurred under circumstances of vast over-expression of the “rescuing” gene.
In a later paper, they followed up on one of the rescuers to see if its new activity could be improved. It was improved about two orders of magnitude, but that left it seven orders of magnitude lower than the wild-type gene it was replacing.
My point? It is easy to propose that there is sufficient substrate or catalytic plasticity to allow the evolution of the protein diversity we see know. That is a hypothesis, and not a demonstrated one.
It’s reasonable to ask what range of activities most enzymes can carry out, or how many functions can be substituted for. But right now it certainly is not demonstrated.
If specificity is continually shifted in small steps then I don’t see why those small steps can’t add up to large steps over time. It’s a bit like saying you can walk to the curb, but you can’t use the same process to get to the store a few blocks down.
It is easy to propose that mutations will knock out beta-lactamase activity, but it is quite another to propose that knocking out beta-lactamase activity does away with any potential activity in a protein. That hypothesis needs to be tested.