2 interesting papers (one a preprint) have been published in the last couple of days on the impact of DNA shape/structure on mutation rates.
Abstract
Approximately 13% of the human genome can fold into non-canonical (non-B) DNA structures (e.g. G- quadruplexes, Z-DNA, etc.), which have been implicated in vital cellular processes. Non-B DNA also hinders replication, increasing errors and facilitating mutagenesis, yet its contribution to genome-wide variation in mutation rates remains unexplored. Here, we conducted a comprehensive analysis of nucleotide substitution frequencies at non-B DNA loci within noncoding, non-repetitive genome regions, their ±2 kb flanking regions, and 1-Megabase windows, using human-orangutan divergence and human single-nucleotide polymorphisms. Functional data analysis at single-base resolution demonstrated that substitution frequencies are usually elevated at non-B DNA, with patterns specific to each non-B DNA type. Mirror, direct and inverted repeats have higher substitution frequencies in spacers than in repeat arms, whereas G-quadruplexes, particularly stable ones, have higher substitution frequencies in loops than in stems. Several non-B DNA types also affect substitution frequencies in their flanking regions. Finally, non-B DNA explains more variation than any other predictor in multiple regression models for diversity or divergence at 1-Megabase scale.
Thus, non-B DNA substantially contributes to variation in substitution frequencies at small and large scales. Our results highlight the role of non-B DNA in germline mutagenesis with implications to evolution and genetic diseases.

Abstract
Single nucleotide mutation rates have critical implications for human evolution and genetic diseases. Accurate modeling of these mutation rates has long remained an open problem since the rates vary substantially across the human genome. A recent model, however, explained much of the variation by considering higher order nucleotide interactions in the local (7-mer) sequence context around mutated nucleotides. Despite this model’s predictive value, we still lack a clear understanding of the biophysical mechanisms underlying the variations in genome-wide mutation rates. DNA shape features are geometric measurements of DNA structural properties, such as helical twist and tilt, and are known to capture information on interactions between neighbouring nucleotides within a local context. Motivated by this characteristic of DNA shape features, we used them to model mutation rates in the human genome. These DNA shape feature-based models improved both the accuracy (up to 14%) and the interpretability over the current nucleotide sequence-based models. The models also discovered the specific shape features that capture the most variability in mutation rates, and distinguished between the most and the least mutated sequence contexts, thus characterizing mutation promoting properties of the genomic DNA. To our knowledge, this is the first attempt that demonstrates the structural underpinnings of nucleotide mutations in the human genome and lays the groundwork for future studies to incorporate DNA shape information in modeling genetic variations.
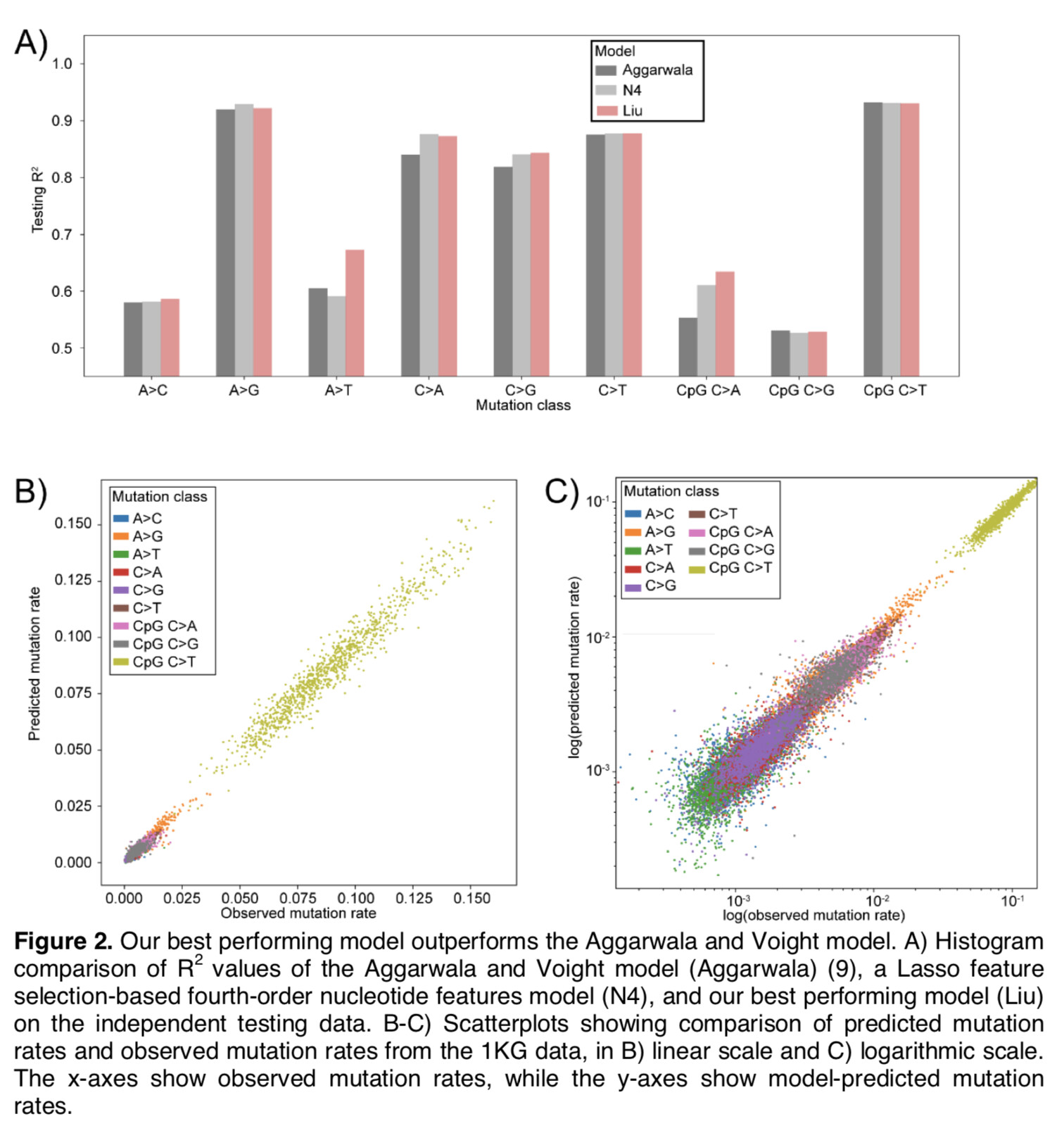
(I may add some more description/details later if I have time, but for now I just wanted to post them for those who might be interested.)