I would have to consider whether that was more a matter of coding for the expression or the actual protein. Your ribs may look much different from your femur, but the materials are essentially the same.
1 Like
Nice use of the passive voice, but they have excluded themselves from not merely mainstream science, but science itself. They don’t test their hypotheses, they just proclaim them to be true.
That’s objectively false, as they have more than sufficient financial resources. Their self-exclusion from science doesn’t prevent them from starting to do some.
From a design hypothesis, yes. Evolution engages in repurposing. You’ve managed to falsify an empirical prediction of your design hypothesis again!
Exactly. Moreover, the same regulatory proteins are reused for different morphologies. It’s amazing that Gil can’t be bothered to look these things up.
1 Like
In his defense, @Giltil wasn’t talking about regulatory proteins. He was talking about junk DNA, which he thinks isn’t junk but a mass of regulatory sequences, mostly RNAs. So your and Ron’s comments didn’t actually address his claim. In particular he was trying to explain the lack of conservation of ENCODE features between species.
I didn’t claim he was. He was assuming that different structural proteins would be used, as would I from a design hypothesis. That’s why I started my second sentence with “Moreover,…”
But they did. Lack of conservation because Gil falsely assumed that different genes are used to produce different morphologies. That assumption is more accurately described as a misrepresented empirical prediction of his design hypothesis. He and Bill do this very often.
No, he wasn’t talking about proteins of any sort. He was talking about the sorts of sequence features ENCODE found and that he assumed were functional in controlling gene expression.
Again, he’s saying nothing at all about genes (i.e. protein-coding genes and structural RNA genes). And again, he’s talking about (mostly spurious) transcription factor binding sites, methylation patterns, and the other stuff that ENCODE looked at. Those are the functional elements he means.
That may be a prediction of the design hypothesis, but it’s not a prediction that @Giltil made.
He stated it much more generally, as “genetic features”:
Again, that’s how intelligent designers would do it, not evolution. But if you want to split hairs and presume that Gil writes about biology with any precision, you’re right and I’m wrong. Can you admit it when you are wrong?
Context should inform you of what we were arguing about just then.
I wouldn’t agree with that either. In either case, different morphologies would require some genetic differences, and those differences could easily be small or large. There really are no expectations of design, particularly when that designer is supposed to be omnipotent.
There’s no need to split hairs and there’s no need to presume precision on his part; context is clear enough. But you need to beat on him for what he’s actually saying rather than what he isn’t.
Sure, when I am.
This is interesting. Could you give an exemple of two allium species with widely different genome sizes not due to polyploidy ?
I am not contesting that conservation is generally a good indicator of function. Rather, I am contesting the idea that non conservation implies absence of function.
Isn’t conceivable that these non protein coding regulatory sequences are less constrained than protein coding sequences?
That makes zero sense. If non-conservation doesn’t imply absence of function, then conservation can’t be an indicator of function. An indicator must be able to differentiate between presence and absence of the indicated feature. Non-conservation is highly suggestive of non-function, even though some non-conserved sequences are functional. You seem unable to grasp any probabilistic arguments.
There’s a difference between “less constrained” and “unconstrained” which you also seem unable to grasp. And in fact some regulatory sequences are less constrained than protein-coding sequences while others are more constrained. Some protein-coding sequences are less constrained than others. What is being suggested to you is that a complete lack of constraint — neutral evolution — is a good indicator of lack of function, while constraint indicates function. Most of the human genome shows no constraint, so we can infer that most of it lacks function. There may be a few functional sequences hiding in there, because they have acquired function recently. But not many. And similarly there may be a few conserved sequences that lack function because they have lost it recently. You can’t hang your whole story on exceptions, though.
4 Likes
See Figure 2 of:
4 Likes
Ok, maybe I can help in this situation. So the scientists are trying to tell you that there are some exceptions to the rule that constraint followed function, the most significant of which is where there is a relatively newly acquired function that perhaps hasn’t even reached fixation yet.
However, you seem to be suggesting the majority of the completely unconstrained portions of the genome that ARE NOT covered by known exceptions are nonetheless potentially functional.
They (we) are trying to help you express to the forum why you say that is, and not getting anywhere, so I will ask you directly:
do you have any justification for the assertion that seemingly unconstrained portions of the genome that are not subject to known exceptions are nonetheless functional, or is it just a matter of “well you never know?”
1 Like
This is the image that @sfmatheson is refering to.
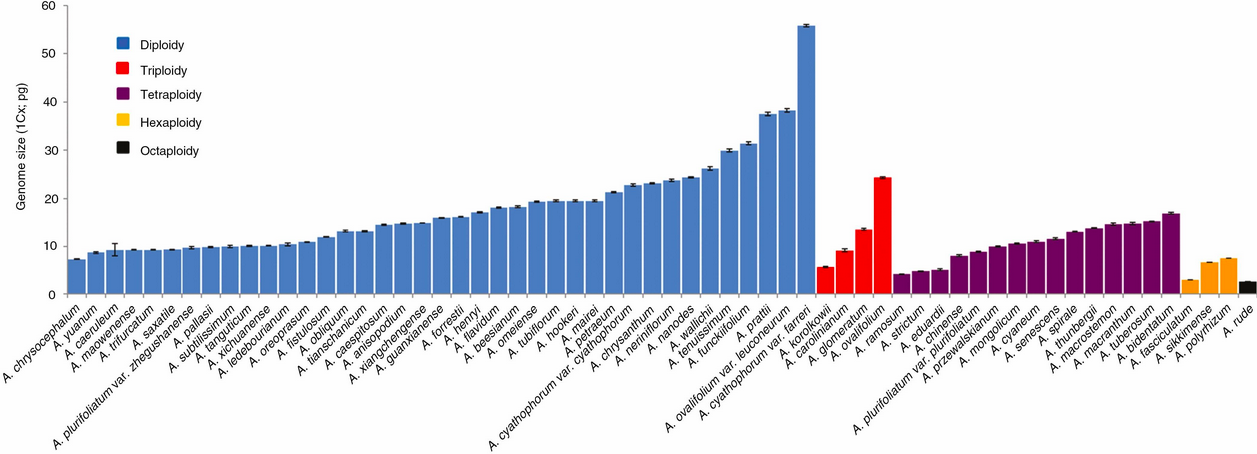
Just to make this clear, the graph depicts the size per monoploid genome (n). This means that the total genomes for diploid (2n) is actually twice as large as is depicted in the graph, and for triploid (3n) it’s three times as large as depicted, etc. So even accounting for the difference in ploidy, there is quite some variation in genome sizes.
A reliable indicator has to have a low probability of false positives and false negatives. Non-conservation, or specifically sequences that evolve according to neutral evolution, is what we would expect of non-functional sequences. If you disagree with that, you need to answer the following question in the next paragraph.
They are STILL constrained, such that these sequences won’t evolve according to neutral evolution; i.e. they are conserved. To repeat myself: you don’t get to discard the absence of sequence conservation when you want to proclaim that most of non-conserved sequences are still functional. You need to explain how a very large part of the genome can maintain their function(s), even if the sequence change in such a way that is exactly as what we would expect from neutral evolution. That’s the question that your position needs to address.
3 Likes
This seems a bit far fetched to me. If I see traces of blood in my home when I return from a long vacation, doesn’t that indicate that someone has broken into my home while I was away? And if I see no traces of blood, can I be sure that no one has entered my home?
What proportion of the genome is under neutral evolution and what evidence do we have that these genomic regions are really under neutral evolution ?
We understand when and where absence of evidence isn’t evidence of absence. This analogy doesn’t accomplish anything towards showing why absence of evidence for a correlation between genome size and coding fraction, to morphological complexity, should be thought of as a point in your favor. You’re just suggesting a relationship not evident in the data and then handwaving in the direction of a principle to explain away why the data doesn’t conform to the relationship you’re positing.
We get that the pattern not being evident doesn’t disprove it’s absence, but that still leaves you with no data in support of your hypothesis.
Approximately 90% of the human genome. Intra and inter-species comparisons in mutation rates shows you what portions of the genome are evolving at and slower or faster than the expected rate for neutrality. If it’s faster it implies positive selection (therefore probably functional), if slower it implies purifying selection (therefore probably functional), and if roughly at the neutral rate it implies no function. There is some grey areas of course, as some functional areas are more or less tolerant to mutations than others, and even for actually nonfunctional DNA we still expect an occasional deleterious mutation that (for example) can create a spurious promoter region that interferes with other cellular processes. But again, to a first approximation it’s a good indicator of the functional fraction.
5 Likes
In school probability is often introduced with examples of dice, coin tosses, marble urns and the like. The sample sizes are kept small for illustrative purposes, the space of possible outcomes is well within a range one can comfortably count. Some weeks ago we saw one consequence of this, where Bill Cole would only define probability in terms of the counting measure, and completely overlook the possibility of non-uniform distributions, let alone non-finite sample spaces. What I’m spotting here with Gilbert is a similar situation.
“Randomness” is something many who do not work with it associate with a form of completely uncontrollable chaos. A random system to them is a system that can pretty much do what ever is within its reach at any point, and there is no characterizing or predicting it. Bluntly put, this is just straight up false. At sufficiently large sample sizes - which, depending on the concrete problem, may be trivially easy to achieve, like in the case of thousands to billions of a genome’s base pairs - “noise” is a grossly insufficient descriptor of any process, not because the process is something other than random, but because of how easy it is to tell the difference between even seemingly slightly different distributions.
Say you are given two dice, one of which is weighted such that one side is, say, 3% more likely than all the others. You will be hard-pressed to accurately and reliably tell which is which after only a dozen or so throws of each, even if you repeat the experiment another time or two. After some 12k throws of each, however, the likelihood to make an earnest mistake in assessing the difference between the outcome distributions is vanishingly small. You know what a fair dice looks like in the high sample size limit. Will every side have an equal share of all the throws? No. But if on the other dice one side dominates the others by as much as 3%, can you honestly argue that this is likely the fair one, and the other weighted?
Much the same argument applies with genome mutations. Surely, something that wildly changes between a handful of generations is likely not survival-critical. We can tell a non-viable organism by how unambiguously dead it is, after all. And this scales down from survival-critical to merely heavily or even slightly impactful. So, if some part of a genome mutates at a rate much different to the other, then the more conserved portion is likely to contain the more important functions. The most mutable portions, on the other hand, must be the ones that either serve functions that have at most a minimal impact on the organism’s fitness, or they serve no function at all. Crucially, we can perform experiments to measure and distinguish those regions reliably. Lastly, we call evolution that has no impact on an organism’s fitness “neutral”. The prediction is that there is (to a first approximation) a positive correlation between how quickly a genome region mutates and the impact mutations in that region have on the carrier organism’s fitness.
It’s not a case of “implies” anything. Correlations are symmetric. If high fitness impact of a region correlates with that region’s conservation, then conservation likewise correlates with fitness impact, and, therefore, low fitness impact correlates with low conservation. Whether there is a causal link, or any other assymmetric sort of entailment, or which direction it points is not something any amount of data can ever reveal to us. We may conjecture such things in the theory, if that helps us understand the model and its implications, but at the risk of introducing confusions, like about whether the converse follows. The data remains what it is, regardless of how well or poorly our models aid us in predicting it.
2 Likes
There you go again, demanding absolute certainty or nothing. Of course you can’t be sure, but would you consider it likely? If you come back a thousand times and see blood once, how many times would you estimate that someone has entered your home? According to you, it should be a thousand.
About 90%, and the evidence is lack of conservationi. This seems rather late in the conversation to be asking such a basic question.
1 Like
Especially since the subjects of neutral theory and genetic load come up within the first screens of Google searches about junk DNA. Well, someone could miss them if they only use the “I feel lucky” button, I suppose.
It seems to me that there are only three cases where lack of conservation is not associated with lack of function. Two of these - gain or loss of function - imply the existence of junk DNA. The third case where all the sequences involved are functional - if considered as near-universal - implies that function is very common in sequence space, contrary to the usual view in ID circles
I don’t see that any of these are helpful to an ID supporter, determined to deny the existence of junk DNA.
6 Likes
The blatant contradiction there is astounding.
2 Likes
From what I have seen, spurious promoter regions seems to be more applicable in prokaryotes. Prokaryotic genomes show purifying selection across the whole genome to minimize accidental promoter-like sequences. Since most of the genomes in many eukaryotes appear to be under neutral evolution, this doesn’t seem to be much of a problem for them. My suspicion is that this has to do with how different the transcription factors work between prokaryotes and eukaryotes. Transcription is essentially active by default in prokaryotes. However, in eukaryotes, even a strong ‘core’ promoter is practically inactive without the help of activators, and efficient transcription often requires the combined effect of multiple activators binding at many sites (associated with distal and proximal promoter) at the same time. Not to mention that densely packed chromatin also silences DNA, even those that contain genes (e.g. X-inactivation). To me, this suggests that the formation of spurious high-active promoters is far less likely to occur in eukaryotes, resulting in a lower genetic load per base pair. On the other hand, while most of the genome of prokaryotes is subject to this selective pressure, they can bear the genetic load due to having small genome sizes, large population sizes, and fast reproductive rates.
Regarding functional areas that are more or less tolerant to mutations. I agree that some regions are more tolerant than others, but to suggest that a sequence can maintain the function(s) even while evolving in a way that is (nearly) identical to neutral evolution would provide the same problem as spurious promoters, but much more severely. How would the cell differentiate such a sequence from the rest of the genome? [e.g. what would happen if any sequence acts as a promoter?] No way to distinguish signal from noise. The only exception I could think “spacer” DNA, where the sequence of ATGC’s doesn’t matter at all, as long as it’s within a certain size range and at a particular loci. Thus function is differentiated by size and location, but even those sequences would be constrained. Perhaps it’s difficult to tell the difference between neutral evolution and non-neutral evolution in such edge cases? I am not sure about that.
1 Like